About Limb-Girdle Muscular Dystrophy
What is Muscular Dystrophy??!!
Girls Chronically Rock was birthed out of the need for community after I was diagnosed with Muscular Dystrophy a few years back. A rare diagnosis, only affecting about 50,000 Americans, I realized many people don’t quite know what MD is. Before my life had to make room for it, I didn’t know what is was either. Let’s answer some of the common MD questions:
https://youtu.be/9ZBwt2dJXwY
What is muscular dystrophy?
Contrary to popular belief, muscular dystrophy is a series of muscle diseases that causes the weakening of the skeletal muscles over a series of time. The patient’s body has a mutation, some abnormal genes start to interfere with the production of proteins needed for the body to generate healthy muscle.
There are no external causes of Muscular Dystrophy. Internally, it comes about when certain genes in the body are mutated, causing the production of proteins in the body to generate healthy muscle fibers.
Once a person has the mutation, MD can become hereditary. Those with a family history of the disease are at a higher risk of developing the disease or passing it along to their children.
What symptoms should I look out for?
According to Healthline, our bodies house more than 650 skeletal muscles. Therefore, Muscular Dystrophy can impact a very large portion of the body in different ways.
Those who have not been previously diagnosed, they may want to check in with a doctor if they have any of the following symptoms:
- Difficulty walking
- Difficulty with use of arms
- Limited mobility
- Difficulty breathing
- Scoliosis
- Heart problems
- Difficulty swallowing
Depending on the type of MD, a person can have a difference in severity, number, locale and onset of symptoms.
What types of Muscular Dystrophy are there?
We currently know about nine types of MD, but there are six that are most common. Each type contains slightly different genetic mutations and are altered depending on the subtype, if applicable.
- Duchenne – The most common type of MD, this type usually occurs in young boys between birth and the age of two.
- Becker – Most similar to the Duchenne type, Becker MD mimic the symptoms but progresses much slower and tends to be milder in nature. Those with this time usually don’t experience symptoms or diagnoses until their mid-20’s or later.
- Myotonic – Those with myotonic MD are majorly affected in their face and neck muscles. They have an inability to relax muscles after contractions and often experience drooping faces and eyelids and necks that resemble swans’.
- Facioscapulohumeral (FSHD) – Shoulders and hips are first to be affected with this type of muscular dystrophy. Shoulder blades tend to stick out like wings when the arms are raised.
- Congenital – This type of MD impacts boys and girls between birth and the age of two. It can vary from a mild disability to a severe impairment.
- Limb-girdle – Onset of this type of MD typically occurs between childhood and adolescent years. Hip and shoulder muscles weaken first, followed by drastic difficulty with lifting the front part of their feet, their arms, and legs.
What about me?
When I was 24, just finishing up my MBA program, I was hit with an onslaught of symptoms. I had increasing difficulty with moving my legs and lifting my arms. Pangs of pain happened more frequently. And I was falling left and right. It was stressful and utterly confusing trying to assess if it was my weight, or maybe something wrong in my brain. It certainly couldn’t have been my age – I hadn’t even made it to my quarter life yet.
Many doctor appointments later, I was diagnoses with limb-girdle muscular dystrophy. Many years and additional doctor appointments past, I’m still unaware of my subtype. Currently, MD doesn’t have a cure, but can be managed with personalized care plans that incorporates medications, breathing apparatuses, physical therapy, home health-aides (to help with feeding, make up application, bathing, etc.), and transport aides. In my years of dealing with this, I’ve been trying to enroll in trials to see if I could gain insight about my subtype and maybe even get closer to helping finding the cure, but to no avail.
Trials are tricky. They can either help solve the problem or cause a slew of others, making the participant even sicker than they were before. It’s risky business. So I continue to trend lightly, making sure to take notes and the advice of my doctors, be transparent and honest with my home health aide and doing what I can to manage my self-care and positive emotions.
(Re)sources: Mayo Clinic, WebMD, Muscular Dystrophy Association (MDA)
People Always Ask, What is Limb-Girdle Muscular Dystrophy???? Well, here is a depth definition of what it is, the symtoms, the different types and more.
What is Limb Girdle Muscular Dystrophy (LGMD)?
- A term used for a GROUP of rare neuromuscular diseases which are inherited and known to cause muscle weakness & wasting.
- The muscles most affected are those closest to the body (proximal muscles), specifically the muscles of the shoulders, upper arms, pelvic area, and thighs.
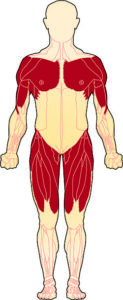
- More than 30 forms of LGMD exist; with new forms (sub-types) being discovered each year.
- Some LGMD sub-types have a ‘dominant’ inheritance & some have a ‘recessive’ inheritance pattern.
- In some cases a family history exists & in others, no family history of the disease is known.
- Onset of symptoms can occur in childhood, adolescence, or even adulthood.
- LGMD occurs in all parts of the world & among all ethnic groups.
- Males & females can inherit LGMD.
- No cure or treatment exists for these progressive diseases although promising research is being carried out which gives hope to many individuals.
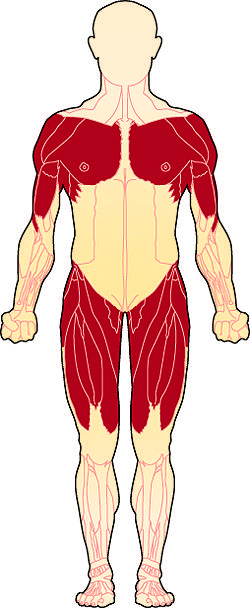
Please learn more information on https://lgmd-info.org/
Also, please feel free to make a donation to the Limb-Girdle Muscular Dystrophy Association. You can do so, by donating here https://lgmd-info.org/organizations/
What is limb-girdle muscular dystrophy?
Limb-girdle muscular dystrophy (LGMD) is a diverse group of disorders with many subtypes categorized by disease gene and inheritance. LGMD usually manifests in the proximal muscles around the hips and shoulders. (The proximal muscles are those closest to the center of the body; distal muscles are farther away from the center — for example, in the hands and feet).
The shoulder girdle is the bony structure that surrounds the shoulder area, and the pelvic girdle is the bony structure surrounding the hips. Collectively, these are called the limb girdles, and it is the observed weakness and atrophy (wasting) of the muscles connected to the limb girdles that has given this group of disorders its name.
What are the symptoms of LGMD?
The unifying features of the LGMDs are the weakness and atrophy of the limb-girdle muscles. However, the age at which symptoms appear, and the speed and severity of disease progression, can vary.
Individuals may first notice a problem when they begin to walk with a “waddling” gait because of weakness of the hip and leg muscles. They may have trouble getting out of chairs, rising from a toilet seat or climbing stairs. As this weakness progresses, the person may require the use of assistive mobility devices.
Weakness in the shoulder area may make reaching over the head, holding the arms outstretched or carrying heavy objects difficult. It may become increasingly hard to keep the arms above the head for such activities as combing one’s hair or arranging things on a high shelf. Some people find it harder to type on a computer or other keyboard and may even have trouble feeding themselves.
Some of the various LGMD subtypes also are characterized by additional symptoms. For example, the heart can be affected in some types of LGMD, with weakness of the heart muscle (cardiomyopathy) and/or abnormal transmission of signals that regulate the heartbeat (conduction abnormalities or arrhythmias).
Some disease subtypes also involve the muscles used for breathing, and for that reason, respiratory function, along with cardiac function, should be monitored regularly.
Other symptoms may be present in some of the different subtypes of LGMD, including but not limited to: joint stiffness, muscle cramps, enlargement of calf muscles and involvement of distal muscles of the body such as those controlling the hands and feet.
What causes LGMD?
Genes are the codes, or recipes, that cells use to manufacture the various proteins needed by the body. The genes associated with LGMD normally encode proteins that play vital roles in muscle function, regulation and repair. When one of these genes contains a mutation (a flaw, such as missing or incorrect information) cells cannot produce the proteins needed for healthy muscles.
There are two major groups of LGMDs. Called LGMD1 and LGMD2, these two groups are classified by the respective inheritance patterns: autosomal dominant and autosomal recessive. If one copy of the abnormal gene is sufficient to cause the disease, it is said to be autosomal dominant; if two copies are needed then the inheritance pattern is autosomal recessive. For more details on the various inheritance patterns, see the NIH Genetics Home Reference.
Dozens of different genes, when mutated, have been shown to cause specific LGMD1 and LGMD2 subtypes. In these cases, the proteins associated with these genes are nonfunctional or deficient, and muscles are unable to function normally. Gradually, the muscles become weak enough that people experience the symptoms of limb-girdle muscular dystrophy.
In addition to the known LGMD1 and LGMD2 subtypes linked to specific genes, there are many cases of LGMD for which the causative gene is not yet known (and the person is not identified to have a subtype-specific form of LGMD). Scientists are actively working to understand the causes of these unidentified subtypes of LGMD, because the more we understand about all the different causes of LGMD and the diverse ways that muscle can be compromised, the better chance we have of finding effective therapies to intervene in the pathological process.
What is the progression of LGMD?
At this time, progression in each type of LGMD can’t be predicted with certainty, although knowing the underlying genetic mutation can be helpful. Some forms of the disorder progress to loss of walking ability within a few years and cause serious disability, while others progress very slowly over many years and cause minimal disability.
LGMD can begin in childhood, adolescence, young adulthood or even later. Both genders are affected equally.
When limb-girdle muscular dystrophy begins in childhood, some physicians say, the progression is usually faster and the disease more disabling. When the disorder begins in adolescence or adulthood, they say, it’s generally not as severe and progresses more slowly.
What is the status of research on LGMD?
MDA-supported scientists are pursuing several exciting strategies in muscular dystrophy research that have implications for LGMD. These strategies include gene therapy, exon skipping, stop codon read through and myostatin blocking.
To learn more, read In Focus: Limb-Girdle Muscular Dystrophy (October 2013 special report).
Subtypes of LGMD
Here is a list of LGMD subtypes. Type 1 LGMDs are dominantly inherited, requiring only one mutation for symptoms to result. Type 2 LGMDs are recessively inherited, requiring two mutations — one from each parent — for symptoms to appear. Sometimes, LGMDs are referred to by their names, not their numbers, and some types have not been assigned numbers.
Some LGMD subtype names:
- Calpainopathy (CAPN3 mutations; recessive; LGMD2A)
- Desmin myopathy (DES mutation; dominant; LGMD1E / DES mutation; recessive; LGMD2R)
- Dysferlinopathy (DYSF mutations; recessive; LGMD2B)
- Sarcoglycanopathies (SGCG, SGCA, SGCB, SGCD mutations; recessive; LGMD2C, LGMD2D, LGMD2E, LGMD2F)
- ZASP-related myopathy (ZASP mutation; dominant; a form of myofibrillar myopathy)
Dominant LGMD subtype numbers:
- LGMD1A (MYOT mutation)
- LGMD1B (LMNA mutation)
- LGMD1C (CAV3 mutation)
- LGMD1D (DNAJB6 mutation)
- LGMD1E (DES mutation)
- LGMD1F (TNP03 mutation)
Recessive LGMD subtype numbers:
- LGMD2A (CAPN3 mutations)
- LGMD2B (DYSF mutations)
- LGMD2C, also called SCARMD1 (SGCG mutations)
- LGMD2D, also called SCARMD2 (SGCA mutations)
- LGMD2E (SGCB mutations)
- LGMD2F (SGCD mutations)
- LGMD2G (TCAP mutations)
- LGMD2H (TRIM32 mutations)
- LGMD2I (FKRP mutations)
- LGMD2J (TTN mutations)
- LGMD2K (POMT1 mutations)
- LGMD2L (ANO5 mutations)
- LGMD2M (FKTN mutations)
- LGMD2N (POMT2 mutations)
- LGMD2O (POMGnT1 mutations)
- LGMD2P (DAG1 mutations)
- LGMD2Q (PLEC1 mutations)
- LGMD2R (DES mutations)
- LGMD2S (TRAPPC11 mutations)
- LGMD2T (GMPPB mutations)
- LGMD2U (ISPD mutations)
- LGMD2V (GAA mutations)
- LGMD2W (LIMS2 mutations)
- LGMD2X (BVES mutations)
- LGMD2Y (TOR1A1P1 mutations)
https://www.mda.org/disease/limb-girdle-muscular-dystrophy